Two books arrived on Sunday, July 7th.
Timothy Gowers’ Mathematics: A Very Short Introduction is a very easy read, covers a variety of topics, including graph theory, and his specific example of graph coloring (pg. 15) nudged me to attempt this blog post.
I am breezily summarizing many published papers here.
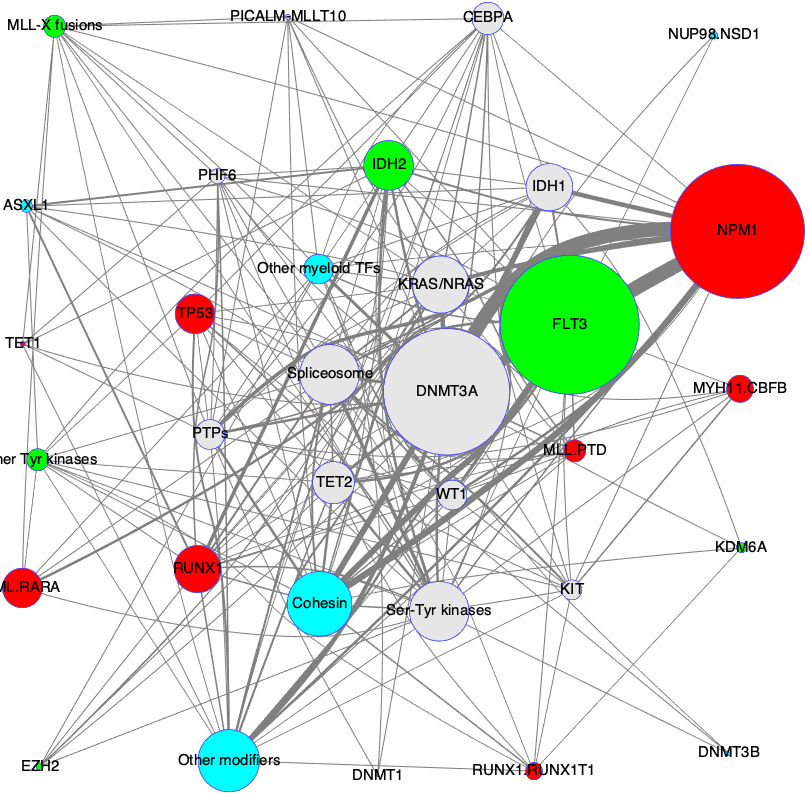
Data sets from The Cancer Genome Atlas (TCGA) are now available, for a variety of cancers, and among other things, include information about which genes are mutated for which patient for any given cancer.
With this, we can create a matrix. The rows of the matrix are patients, the columns are the genes, and the elements of the matrix (row i, column j) are zero or one (so this is a binary matrix), where “one” in (row i, column j) means that gene j is mutated for patient i.
See, we have mapped cancer genomics to mathematics.
Some more biology. Not all mutations matter. The mutations that do not matter are called passengers. Those mutations that matter are called drivers. Not surprisingly, we want to isolate drivers from passengers. This is not easy. Most mutations are passengers.
Even more biology. The same cancer can manifest itself due to different driver mutations.
Why? Because different mutated driver genes can impact the same cellular signaling and regulatory pathways. This mutational heterogeneity complicates efforts to identify functional mutations (another name for drivers) solely by their frequency of occurrence.
This means that we should search at the level of pathways and not just stay on the level of individual genes.
Thankfully, some pathways are already known for some cancers, from parallel research, and searching within them can help find significantly perturbed driver mutations. This is not the hard part.
However, many pathways are still not known.
A key question being asked is this: given the enormous data now available from TCGA, is there an algorithmic procedure — that relies almost entirely on this data – that can “automatically” (that is, with little to no knowledge of cancer biology) identify candidate mutated driver pathways?
We are a support function to cancer researchers and treatment developers. The hope is that outputs of these algorithms provide useful complementarities and enhancements to their own efforts.
To automatically identify useful candidates from data, we need more biology to help us narrow the search for pathways. It turns out, two simplifications can be made, as a first approximation, which leads to a mathematical statement that is recognizable by graph theorists, and so by computer scientists and operations researchers.
To be absolutely clear: a pathway is a collection of genes. To find k pathways means finding k different collections of genes. Each collection of genes can be of various size. You can imagine that the number of such collections is impossibly large.
Thus, the need for some simplifications, leading to a first-pass “combinatorial model”.
Simplification 1: A pathway has at most one mutated driver gene. This is because driver genes are alleged to be quite rare. Thus, two different pathways will not share a driver gene. This is called (mutual) exclusivity.
Simplification 2: A pathway should apply to many patients. This is called coverage. The important thing to note is that even though two patients share a pathway, they can have a different mutated gene from that shared pathway as an explanatory reason for their cancer.
With these two simplifications, when mapped to the binary matrix of genes and patients, the search for pathways sounds a lot like an independent set or graph coloring problem (from Timothy Gowers book).
These problems are computationally hard (in the worst-case complexity sense of theoretical computer science), but reframing them as binary non-linear programs is an option one can take to see how long actual instances take to be solved exactly, or approximation algorithms can be constructed.
This brings us from biology, through matrices and graph theory, to operations research approaches for computationally solving numerical instances.
But working in cancer biology is trickier than that! Why?
The real data is noisy.
Because of measurement errors and passenger mutations, we cannot impose exclusivity as is. Instead, we should allow for some overlap or “approximate exclusivity”. Thus, Simplification 1 needs modification. Similar considerations force a modification of Simplification 2 as well, in the sense that we now can only reasonably hope that most patients have at least one mutation in a pathway.
How to rigorously quantify these needed modifications? Clearly, allowing more overlap and relaxing how many patients can be not covered, leads to finding more candidate pathways. But, are these pathways for real?
This brings us to asking questions about the shape of data, and this is where topology comes in.
A formal way to discuss how much overlap and non-coverage to allow is to ask whether the homology of the data is changing as we change the amount of modification.
An invariant of a topological object is given by its Betti numbers. Two objects are homotopic if their Betti numbers are the same. So we can compute Betti numbers for each amount of modification, and stop further modification when the Betti numbers have stabilized.
This is called persistent homology.
Now we are (finally!) ready to build a model, with just the right amount of modification for exclusivity and coverage, seeking to extract mutated driver pathways from TCGA data about patients and their mutated genes.
Our model has far fewer binary variables (that are linearly constrained) than others in the literature, but it has a non-linear non-convex objective function.
How to solve this as commercially available optimizers do not perform well on such models?
Using our GAMA (Graver Augmented Multi-seed Algorithm)!
And, what do we get?
We tested our approach on two cancers: Acute myeloid leukemia (AML) and Glioblastoma multiforme (GBM).
In both cases, (a) we recovered pathways that other researchers had previously found by other models and computational methods, and (b) found new ones!
It is time for cancer biologists to weigh in and evaluate whether these new candidates are worthwhile to pursue.
Can we do more than find new mutated driver pathways?
Yes.
We can look at the collection of these pathways and ask whether they have a certain pattern as a group.
This takes us deeper into the world of algebraic topology, where a pathway maps to a simplex, and by studying the simplicial complex, we may learn something about cancer in an integrated manner.
What have we found so far? The space of AML pathways is homotopy equivalent to a sphere (genus-0 surface) while that of GBM is to a double torus (genus-2 surface). This suggests that we may be able to classify various cancers by the shape of their space of pathways.
Maybe Ed Sheeran will write a song about it.😏I enjoyed him playing himself in the cute movie, Yesterday, which was way more interesting and enjoyable (love The Beatles’ songs) compared to Spiderman, the two movies that I watched over the July 4th weekend.
Back to cancer biology. Can we do even more? Yes, we can use additional information available from other sources, such as expression data and chromosomal copy numbers. It is possible to also work at subgene level.
And there is the immune system to include in our analysis.
The second book I received, is a “Three volumes bound as one” English translation (from the Russian) titled:
Mathematics: Its Content, Methods and Meaning.
This book, surprisingly readable 🤷🏽♂️, can be viewed as a “Mathematics: A Ridiculously Long Introduction”!
Why read these kinds of books?
Because it may further help me bring The Language of The Gods to the World of Men.